


The Alchemy of Metabolomics: Erythritol
Headlines on the harms of 'artificial sweetener' erythritol broken down


As the media wanes on caring about real pandemics, it seems we are back to nutrition/metabolism science generating clicks about *potential* harms. This edition of media *maybe* panic is about a new investigation in Nature Medicine on the sweetener/metabolite erythritol and cardiovascular disease (CVD). Let’s dig in:
First - the study comes from Stanley Hazen’s group. Hazen is notable for having launched the TMAO-CVD link back in 2011 and 2013. In many ways, it mirrors what’s happening in the current investigation, employing untargeted metabolomics to plasma samples from large cohorts of humans, looking at what is associated with chronic disease and highlighting that the circulating metabolites have some relevance to dietary intake. This somewhat naturally leads to the implication that maybe changing diet would be therapeutic, modifying related dietary variables to modify the metabolite linked to disease. This is a fine approach and generates nice hypotheses capable of being tested, but needs to be interpreted carefully, and as is often the case, one paper doesn’t typically have the level of evidence necessary to resolve uncertanties in science enough to change dietary recommendations despite enthusiastic authors/institutions/media organizations/influencers wanting you to.
Let’s dive into the specifics of the current erythritol paper a bit more:
The group utilizes untargeted metabolomics data (measuring many metabolites at once and attempting to resolve their chemical identities) generated from the GeneBank cohort at Cleveland Clinic. This cohort has been mined quite a bit for metabolite-disease relationships and you may recognize it as the same cohort that uncovered the TMAO-CVD link. In this paper, baseline untargeted metabolomic data from 1157 individuals samples were analyzed in relation to major adverse cardiovascular events (MACE) over the following 3 years; among the top hits associated with MACE were polyols. Polyols are sugar alcohols that have shown up in human bodily fluids (plasma, urine and CSF) throughout the history of the study of metabolism and have been on occasion noted to be altered in disease states (earlier discussions of these in human metabolism, apart from safety data around them as sweeteners, focused on rare inborn errors of metabolism). Some can be found in the diet at naturally low levels or added as a low calorie sweeteners, and probably some are made by the microbiome, but the several major polyols found in the body arise from the pentose phosphate pathway (PPP). The PPP is a major pathway where glucose is shunted towards production of ribose 5 phosphate, the building block for nucleic acids, and NADPH, needed for several functions including fatty acid & amino acid synthesis as well as glutathione generation (the cell’s major antioxidant defense system). In this metabolomic analysis, erythritol as well as several other polyols including xylose (a 5 carbon sugar found in cereal grain fibers also used in medical tests for intestinal malabsorption and similar in structure to the sweetener, xylitol ), threitol ( a human metabolite of xylose) and arabitol (a major yeast metabolite) are top significant metabolites associated with having a MACE event vs not. Other intesting metabolites show up as well, such as myo-inositol (increased in MACE vs not) - an important secondary messenger within the cell and component of membrane lipids, and also a popular supplement.

The authors breakdown risk by quartiles of erythritol and really only see an meaningful increase in risk of MACE and death in quartile 4 (the highest levels of erythritol). At this point, it’s worth noting a couple things: 1) we don’t know if the erythritol is there because they’ve eaten erythritol or because of what was made naturally in the body (more on that later); 2) we’re looking 3 years out - so folks who are having a MACE and/or dying within 3 years of their blood draw used for metabolomics are pretty sick at baseline. The authors see modest attenuation here adjusting for other risk factors (including age, sex, diabetes, systolic BP, BMI, LDL and HDL-C, triglycerides and ‘current’ smoking status). We could also quibble here that other major risk factors, like impaired kidney function weren’t adjusted for, a common cause for some metabolites appearing high in at-risk cohorts (impaired excretion of the metabolite).

In untargeted metabolomic investigations, it’s best to try to reproduce metabolite-disease associations in validation cohorts. This paper is rigorous in that it goes on to look at targeted analysis of erythritol-disease relationships in 2 others - non-overlapping samples from the GeneBank cohort and in the German LipidCardio study. Both cohorts also had 3 years of follow up and had a suite of additional risk factors to consider. The validation cohort data is below and largely repeat the discovery cohorts findings, though the adjusted analyses show significantly more attenuation of risk. The targeted analysis here is helpful because it allows us to get absolute quantification of erythritol levels; untargeted metabolomics isn’t great at estimating true absolute values of metabolites in circulation and often relies on comparing relative values from the area under the curve of metabolite peaks but with targeted analysis we can utilize erythritol standards and an isotopically labeled erthritol internal standard to get at the absolute values. These numbers are shown in the extended data figure 3 and unsurprisingly, had to be log transformed because the data is skewed (consistent with only Q4 but not Q1,2,3 having an association with disease risk). The majority of the samples range between log 1-3uM Erythritol with the distributions substantially overlapping for MACE vs no MACE. I’m not entirely sure if this Figure (extended data 3) is accurate either , given that they are reporting log (Erythritol uM) as the Y axis but reporting values less than 1, which would be transformed back into negative uM values - that can’t happen. This might be a Y axis labeling error issue or some mathematical wonkyness that isn’t clear but what is worth noting is that the values are in the very low micromolar range - old reference ranges for ‘normal’ serum erythritol you can find in the inborn error of metabolism literature report that controls expect to have <5uM.

The risk of a MACE (comparing Q4 vs Q1 plasma erythritol levels) was also looked at amongst patient subgroups (stratified by various risk factors/metabolic values) and we see some interesting factors that appear to modify the risk association. Unsurprisingly, the Q4 vs Q1 comparison shows much higher hazard ratios for folks with impaired eGFR - we don’t get the raw values of what Q4 and Q1 are for these subgroups but it’s likely that with impaired kidney function, the differential between Q4 and Q1 is much greater, consistent with impaired kidney excretion of erythritol. In the European cohort, the diabetes (yes/no) subgroup also stands out with a significant hazard ratio differential, pointing possibly towards both increased production of erythritol and/or impaired excretion. It’s worth noting here that it’s known that erythritol levels are associated with central adiposity and increased in plasma in response to oxidative stress. That info coupled with the variation in erythritol levels by risk factors and clinical covariates, i think we can can pretty fairly say that the erythritol signal we’re picking up on here is from endogenous production and impaired excretion, not from meaningful differences in intake.
At this point, there’s not much to say that erythritol isn’t just a biomarker of disease that’s further capturing risk of having a MACE beyond other traditional risk factors. Whether erythritol itself is causally impacting disease, or is just a marker of metabolic dysfunction and impaired kidney function can’t readily be determined known from these epidemiological cohorts and a certified epidemiologist would likely call out the smaller #’s of individuals in these cohorts (1000s, not 10s or 100s of 1000s) and the relatively unsophisticated modeling/adjustment approaches here. Indeed, adjusting for a few known risk factors like age (Q4 of plasma erythritol was older) as well as other metabolic risk factors and seeing significant attenuation always leads to the concern that the residual risk is explained by imprecisely measured risk factors or a few unmeasured risk factors. Alternatively, it stands that erythritol could be on the causal pathway by which some factors might be biologically influencing risk. These cohorts really aren’t large enough to start to do this level of exploration in the data and confidently say much about whether erythritol is just highly colinear with existing risk factors, mediates the effect of other risk factors or a truly novel risk factor on its own.
Given the uncertainty in interpreting the cohort data, the authors go on to do in vitro and in vivo assays to assess whether erythritol has functional signifiance by looking at platelet aggregation (a theoretical risk factor for thrombosis potential/in vivo heart attack and stroke). Essentially, these assays assess aggreggation of platelets in platelet rich human plasma after stimulation with ADP or TRAP6. Preincubation with relatively high concentrations (compared to our epidemiological cohort concentrations) of erythritol (~45uM) pretty consistently increased platelet aggregation and activation phenotypes tested but no effect was seen with just glucose or another polyol 1,5 anhydroglucitol (though this marker was negatively associated with CVD events in their study). The authors bolster these findings with whole human blood exposed to a collagen coated microfluidics device looking at platelet adhesion, again showing 45uM of erythritol worsens platelet adhesion. In a mouse model of arterial injury induced by iron chloride where blood flow is expected to be stopped due to a clot, pretreatment with a high dose of erythritol (25mg/kg achieving 290uM erythritol) reduced the time to stopped blood flow, indicative of more rapid clot formation.
The levels of erythritol used in these in vitro and mouse experiments are pretty high - much higher than the normal <5uM range seen typically in humans and seen in the original discovery/validation cohorts. To suggest relevance of these experiments to humans, the authors rely on ‘the first phase’ an ongoing non-randomized study of xylitol and erythritol consumption (30g in a single oral dose; 10 packets worth of truvia’s) designed to assess platelet aggregation. This study aims to recruit 40 individuals but in this paper, we get 8 participants who the authors note are not part of that total 40 - essentially, we are getting the authors’ pilot data that was not pre-registered but is anchoring on a different pre-registered trial because its hard to get human intervention data published nowadays without a pre-registration. The authors report that the plasma levels are low at baseline (3.27-4.14uM) but rise to 4.3-7.68mM (an order of magnitude higher) for the first 4 hours after ingestion, and stay elevated above 18-45uMs (thresholds derived from their in vitro studies) for over 2 days. No functional outcomes are reported, just erythritol concentrations.
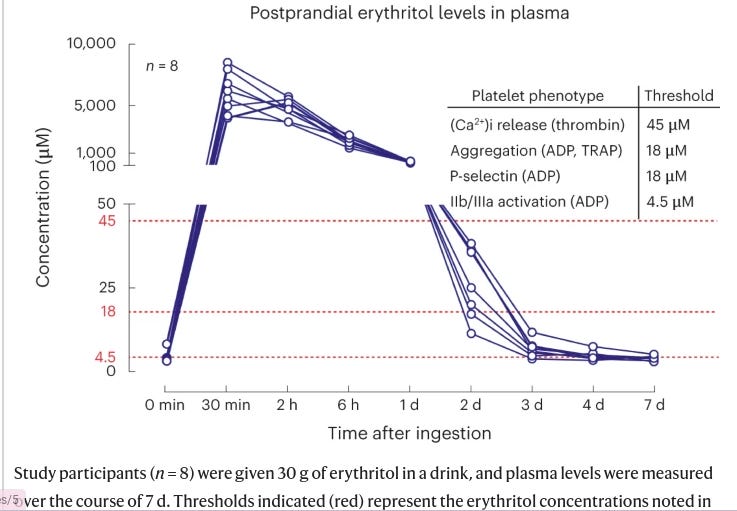
What do we make of this? There’s a few things worth highlighting/discussing.
The purpose - a lot of the media coverage here has been about erythritol, which is scarily labeled an artificial sweetener now, even though its historically been referred to as a natural sweetener juxtaposed against actual artificial sweeteners. It exists in nature, so its not artificial, but the amounts we are consuming in drinks is relatively novel. If you look at this study as a cool use of metabolomics to look at markers associated with disease, and some very preliminary follow-up worth more investigation, that’s great. If you see this and think, it’s time to scare everyone away from using any erythritol ever, you might be putting the cart before the horse and probably just selling clicks or bolstering a fearmongery wellness brand that has folks micro-optimizing every aspect of their lives.
Disparate lines of evidence - the study gives the appeal of having multiple epidemiological cohorts, in vitro mechanistic data, and a human trial that might lend itself to the idea that erythritol is indeed worrisome - how could you argue against so many lines of data?? It doesn’t take very close inspection to realize that these lines of evidence are not all that complementary, despite one investigation having inspired the next. For starters, the epidemiological cohorts are comparing fasting blood levels of erythritol (a pretty narrow range of low micromolar concentrations) with disease risk. These cohorts didn’t measure erythritol intake (hard to do accurately) and its extremely likely the differences being detected here represent endogenously made erythritol and to some degree, a failure to excrete it in the urine. While these cohorts inspired the in vitro data, we see pretty clearly that the differential in circulating erythritol in human cohorts (a few uM) is not what’s being modeled in the in vitro data - these are comparing 0 erythritol to commonly 18-45uM+. Dosing aside, the relevance of these assays to human disease is pretty unclear. There are some very small subcohorts of larger cohorts that suggest ‘hyper-responders’ to ADP induced platelet aggregation (>50% maximal aggregation) have a slightly increased risk of CVD - unfortunately, these assays aren’t always standardized (e.g. different platelet rich plasma preparation protocols, amounts of ADP, different time courses) to allow comparing across studies (indeed, they’re often meant to look clinically for obvious platelet aggregation defects), don’t have standardized risk thresholds established for predicting CVD events, and haven’t been readily assessed in the context of chronic disease risk assessment for environmental variables that increase ADP-induced aggregation ex vivo (contrast this with established surrogate risk factors like blood lipids and blood pressure that readily allow for clinical risk assessment). I’ve talked with some friends in cardiovascular physiology research who note that these assays are pretty promiscuous, in that a lot of things aggravate ADP-induced platelet aggregation, and you’re left with little idea how to interpret it. To add some additional context to these assays, it's notable that arachidonic acid (ARA), an omega 6 fatty acid, can be used as the stimulus to cause platelets to aggregate - you might think, hey, ARA can be eaten in the diet and is the end product of the common dietary precursor, linoleic acid. One could conclude that eating ARA and LA would then lead to increased platelet aggregation - but nutritional physiology is complex, and ARA/LA in human trials don't
Lastly, the human trial turns towards explicitly elevated erythritol through dietary means (unlike the cohorts which is very unlikely to be through diet) and here we achieve very acutely high levels of erythritol in plasma, an order of magnitude higher than the cohorts and much higher than what is seen in the in vitro and animal studies. This is achieved through very abnormal dosing of erythritol (30g in one sitting), not mimicking human intakes but not an intake level that’s impossible to achieve either. I think the biggest shame here is that Nature Medicine allowed for this un-pre-registered pilot data to stand in for the actual clinical trial data, which would’ve told us more interesting information about whether this dosing from erythritol is clinically risky or not. Trying to extrapolate from in vitro data to these clinical levels is challenging - you could both argue that the in vitro data has limited meaning, or you argue that it does have meaning and “OMG! look how much higher the human intervention study’s blood levels of erythritol get than the thresholds in the in vitro assays!!”. You wouldn’t be wrong for thinking maybe these millimolar concentrations of erythritol are extremely risky relative to the high micromolar concentrations in the in vitro assays - but fortunately, tucked away in the extended data figures we get to see that the authors did look at mM concentrations of erythritol in vitro and, while higher than the uM concentrations, it is not anywhere close to a 100-1000x increase in concentration=100-1000x increase in potency. Instead of a ~1.25-1.5X increase in aggregation, 2-3X increase in aggregation. these assays look like they were done at different times (given the double vehicles/0 erythritol lanes) so a caveat here is that direct comparisons may be challenged a bit but it doesn’t appear that the order of magnitude change introduces a similarly substantial change in aggregation.

What to make of this? Alas, we are left to string together disparate lines of evidence and decide whether the collective picture is compelling enough to pre-emptively stop consuming or limit erythritol intake.
You might be thinking to yourself - how did erythritol get approved? There’s actually a reasonable amount of animal toxicology data on erythritol that lead to its approval for use as sweetener globally (WHO, FDA, EFSA all approve). In humans, erythritol is commonly presumed to be beneficial due to its replacing sucrose sweeteners as it is relatively neutral on chronic disease risk factors that sucrose-sweetened beverages/products tend to worsen - granted, there are few chronic studies and the little data you can find is confined to pilot trials. The potential that erythritol has unique impacts on human CVD risk is not impossible of course, and the potential that it raises risk for thrombotic events would be unlikely to be picked up in animal models that are at very low risk of ASCVD (much animal toxicology data is focused on gross/histopathological abnormalities, cancers and basic serum chemistries). It will be interesting to follow this space as clinical trials inevitably get launched and published but for now, the results require you to place a ton of faith in some in vitro assays of questionable relevance. One interesting point to consider for ‘evidence-based’ guidance is that this will be an area where we don’t have much nutritional epidemiology, given how poor dietary assessment tools are at capturing the specific types of non caloric sweeteners added to foods and the fact that these products regularly have their formulations altered (i’ve seen erythritol both added and removed); it might be possible to capture high levels in fasted blood in large cohorts given the reported 2-day period of elevated high concentrations seen in the small trial component of this Nature Medicine paper but it’s unlikely that you’ll have many people consuming enough erythritol at high enough doses to get a large enough sample size to adequately detect an association with chronic disease risk. Thus, we’ll probably need a blockbuster human trial demonstrating an impact of feeding nutritional doses of erythritol on some sort of meaningful surrogate indicator of cardiovascular risk to move the needle of saying erythritol is unsafe.
Fortunately, I get few questions about erythritol in my clinical practice and don’t like erythritols cooling sensation myself so i won’t have to make too many decisions based on this new data. I suspect there are many food processors and formulators sitting down to decide whether they move erythritol out of their products and choose one of the many other sweeteners out there on the market - it only ever made it into product formulations because it was a ‘natural’ sweetener and i’m sure there are food chemists with big ‘WTF’ letters across their foreheads reading these headlines calling it artificial. With the new classes of stevia and monk fruit based sweeteners out there, i’m not sure erythritol is really all that necessary for food manufacturers, apart from being a nice, GI-friendlier sugar alcohol to replace sugar in baking blends. Curious to see the market response here.
As an aside, I was a PhD student at Cornell when Kate Hootman, PhD, RD was wrapping up their dissertation that included an epidemiological analysis that was one of the first to stumble onto erythritol being elevated in folks with elevated central adiposity and showed with some collaborators doing isotope tracing that erythritol is made from glucose through the PPP. Dr. Martha Field at Cornell has continued to do work in this space, identifying alcohol dehydrogenase 1 (ADH1) and sorbitol dehydrogenase (SORD) as being the enzymes capable of reducing erythrose in the PPP to erythritol, showing that oxidative stress upregulates erythritol synthesis, and further assessing the impact of chronic exposure in mice in the context of a high or low fat diet on body weight gain and glucose tolerance (no significant effect). The Field lab also has a recent preprint out showing that mice consuming high sucrose concentrations in their water have elevated erythritol in the urine and plasma, indicating that plasma levels could be a marker of simple sugar intakes as well. This is a lab to watch if you’re interested in following some of the science on erythritols health effects.
A final note to conclude on - expect to see more of these types of headlines. Metabolomics investigations are very common these days, and linking a metabolite associated with disease to diet in some way is extremely easy (typically only a degree or 2 of separation away at max). We will have abundant ‘metabolite related to diet associated with disease’ headlines that we have to sift through - we should always be asking for whether there is direct evidence, ideally in humans, of harm from intake. Erythritol is a great example of something that is in the diet but can also be made endogenously, where we are not sure if it is causally linked to disease but there is speculative evidence to say that it might be, so the media and influencers will run with headlines about it. In isolated incidents this might sound reasonable, but when we ultimately look at a body of evidence, full of associations in every which direction and speculative mechanistic data that points towards changing our diets in some way, we’ll inevitably just reinforce the current state of affairs in nutrition where we have 50 Netflix documentaries claiming wildly disparate diets are the best for you because of *handwaving*.