
Keto, LDL-C, & LMHR, Oh My!
The curious case of Keto-CTA

A new publication in the Journal of the American College of Cardiology titled “Plaque Begets Plaque, ApoB Does Not: Longitudinal Data From the KETO-CTA Trial” has caused a bit of a storm on social media in the nutrition and cardiology space. This cohort study is being hailed as both definitive evidence of risk as well as no harm from the increase in LDL-C commonly seen on a ketogenic diet. This paper as well as the whole topic of keto and LDL-C has an interesting history. Let’s dig into it.
If you went back in time to 1920s and told Russell Wilder and Mynie Peterman that their pioneering use of the ketogenic diet to treat epilepsy would stand the test of time and still be used for that indication to this day, I suspect they might be quite proud. I’m not sure, however, how they’d feel learning that there’s an online diet community who believe that a low carbohydrate's diet effect on blood lipids has no relationship to cardiovascular disease risk (tbf, i’d first have to explain LDL-C and reddit).

Back in the 1920s when keto was being pioneered for the treatment of epilepsy, there were few options for the condition, apart from pretty extreme fasting. Given that ketogenic diets could relatively liberalize the diet and be used long-term with the risks of starvation, the ketogenic diet was a pretty life-changing therapy and its side effects were minimal relative to its obvious benefits and the alternatives. The effect of the ketogenic diet could be readily observed in patients (the dramatic improvements in seizure control being more obvious and less prone to reporting bias) and it was largely considered “evidence-based”, being employed by physicians Samuel Livingstone and Lydia Pauli as well as dietitian Millie Kelly for 4 decades (1930s-1970s) at Johns Hopkins. The KD would lose favor as sodium valproate became a widespread treatment in the 1970s until 1998, when NBC’s Dateline aired a story of a 2yo boy named Charlie who was treated at Hopkins by the KD. The Charlie Foundation was formed and the use of the KD increased dramatically again.
The use of the KD for epilepsy was focused on medical management of a chronic condition that rapidly manifests. There was little investigation into the impacts of the KD on metabolic homeostasis until the 1960s, when a new focus on blood lipids emerged as a modifiable risk factor for cardiovascular disease and new methods to measure blood lipids emerged. Researchers at NIH began assessing blood lipids in children before initiating the KD and after being on it for a sustained period of time, and noted increases in lipids. The KD being used at this time is the traditional one, defined by the ketogenic ratio (most commonly 4:1) - this means that there are 4g of fat for every 1 g (carb+protein). The modern KD is often seen as high protein but protein is anti-ketogenic, similar to carbohydrate, and excesses of it beyond the requirement for essential amino acids has always been historically restricted in the KD. Because fat is >2X as energy dense as carbs and protein, a 4:1 ketogenic diet ready become ~85% of total calories from fat, typically 10-12%E as protein and minimal carbohydrate.

The KD went outside of the clinic and more mainstream for adult populations with the Atkins diet, a low carbohydrate diet that starts off with a very restrictive KD in the initial phases (granted more liberalized on protein). Atkins published his first book in the late 60s but it didnt become super popular til its 2002 rerelease of the “New Diet Revolution”. In both its first release and more modern editions, the diet received pushback for being high in saturated fat and dietary cholesterol, linked to increased blood cholesterol levels.

Despite decades of concern about low-carbohydrate diets and increased blood cholesterol levels, we really don’t know why this happens. Many assume it’s because of the very high saturated fat content (for context, we often debate about the utility of reducing saturated fat intakes from current 11%E down to <7%E; a KD can readily get to >40% kcals from saturated fat). Despite this concern, there’s only a little bit of data modifying fat composition in the context of a KD from short term trials as well as some retrospective analyses of clinics that switch their formula composition. This data lends some support for the idea that fat composition is a major effector of the average increase in LDL-C seen in patients but may not be the only thing. The KD, for example, may be increasing endogenous cholesterol synthesis rates in some or its disposal as bile that are independent of hepatic clearance of LDL-C; other factors like very low viscous fiber and high dietary cholesterol intakes on the KD might have some effect here.

The studies suggesting fat composition drive an increase in LDL use strict 4:1 KD’s based predominantly on formulas, not the everyday version of keto that is typically carbohydrate restricted and protein liberalized that don’t achieve the same level of ketosis. In the everyday person randomized to just a very low carb diet through food and not protein restricting, where we don’t have control over what the diet is and there is often weight loss involved, you see wide estimates of the diet’s effect on LDL-C reported. An emerging hypothesis to explain this variability is that lean individuals respond with greater changes in LDL-C when consuming low carbohydrate diets than do individuals with obesity - a phenotype that is being referred to as the lean mass hyper responder (LMHR). The hypothesis more or less attempts to state that individuals who are lean and consume a very low carbohydrate/KD-type diet are more likely to experience very high increases in LDL-C, moreso than those who are not lean. The seminal meta-analysis suggesting lean individuals’ LDL-C does hyperrespond to a low carb diet has a bunch of flaws but i’m not opposed to the idea entirely. We’ve known for a while that individuals with anorexia nervosa also exhibit a hypercholesterolemia - a disease state dissimilar from the KD in many ways but having a shared metabolic state where the body is burning/relying on fat for energy, suggesting something about burning fat can alter lipoprotein metabolism to increase LDL-C. We’ve also known for a while that the effect of saturated fat on LDL-C isn’t as strong in individuals with obesity relative to a lean phenotype. The identity of a true LMHR phenotype at large is questionable without more representative data but the potential for heterogeneity in the LDL-C response to be explained by something about adiposity/leanness is not outside the realm of possible.
The ‘why’ of LDL-C increasing with a low carbohydrate diet and whether this is due to leanness is only half the battle and brings us to the current publication today. Whether this LDL-C increase is ‘bad’ has come into question from a subset of activists in the low carb space. The reasons for doubting LDL’s causal risk relationship to atherosclerotic cardiovascular disease are myriad but rarely hold up to scrutiny. LDL-C levels are undoubtedly linked to an increased risk of atherosclerotic cardiovascular disease across the strongest study designs we have and highly predictive of risk. Arugments against this follow a few trends. They will cite single measures of LDL-C, commonly in patients who just had a heart attack, where LDL-C is not different between those who had and didn’t have a heart attack and say, A-HA! see, LDL-C didn’t cause the disease. Of course, the LDL-C is measured after the heart attack, precluding any causal inference, and there is a known effect of LDL-C being reduced by acute stresses like a heart attack. Single measures of LDL-C are often invoked in outlier studies, and these fail to capture LDL-C exposure across a lifetime, the real risk we are worried about.
In the low carb space, apart from denying any causal link between LDL-C and ASCVD, advocates and associated researchers have decided that something about the metabolic effects of the low carbohydrate diet obviate the risk from the elevated LDL-C. This can be seen in Dave Feldman’s ‘lipid triad’ model purporting that LDL-C isn’t a problem if HDL-C is high and TG are low - an entirely speculative model that is not supported by available data. Folks have also claimed that LDL-C just isn’t harmful when you’re keto, for less clear reasons. I can be generous and say maybe there’s some anti-inflammatory effects of being in nutritional ketosis that might modify the risk but this is also speculative and needs hard data.
The obvious place to consider is mouse models, which are of course limited as they are not little humans, but nevertheless can provide biological plausibility. Unfortunately, the limited investigation we have doesn’t show a magical effect - the KD isn’t quite as bad as an obesogenic/metabolic syndrome-inducing diet but it’s much worse than control mice who are fed low fat diets that are healthy for a mouse. This data goes against a protecive effect of keto and actually runs counter, indicating keto can worsen increase plaque buildup when blood lipids are high.

Beyond mouse studies, you’re at a bit of a loss. Typically, we would test nutritional hypotheses in a large cohort study where we’ve asked people about their diet or use a biomarker of dietary intakes and follow them until they have a cardiovascular disease event. In most cohorts, few people are intentionally following a ketogenic diet. Folks who are reporting very low carbohydrate diets are commonly doing so because they have overall low intakes or are making diet changes because they are at high risk of disease, increasing the risk of reverse causation. You can’t measure ketones in blood/urine and readily infer nutritional ketosis given that ketones increase in a number of disease states. You need essentially a totally new cohort.
When I was a postdoc, I vidily remember having calls with Spencer Nadolsky, a physician and friend of mine who has been an advocating for nutrition and lifestyle approaches for a while now. He had seen a big increase in folks going keto and their LDL-C increasing, knew the lack of a relevant cohort and saw the emerging arguments that asserted that high LDL-C isn’t harmful if you’re in keto. He had connected with Dave Feldman of the lipid triad-type hypothesis who wanted to get a cohort going. In principle it’s pretty simple - you have a growing group of individuals following a specific dietary pattern online who are organizing into forums & online spaces, and you want to study their risk of atherosclerosis. The realities of establishing cohorts are tough though, especially for folks who exhibit an elevated risk factor for a disease - it’s expensive, it needs ethical approval, you need a hosting institution with an IRB who will oversee data collection and management, you need to collect broad histories on people’s diets, blood lipids & other labs, weight status, medication use, medical histories. You’ve gotta decide on inclusion and exclusion criteria in the cohort which can massively affect sample size & rate of recruitment. You’ve got to pick a primary outcome to measure in the cohort that is reflective of disease risk, estimate the impact of the exposure (high LDL-C) on that outcome and adequately power the cohort (come up with a sample size) to detect whether there is a clinically meaningful effect. All of this is normal for a cohort but typically done with large and well-trained academic teams. Adding complexity here, you’ve got folks online who are reporting LDL-C’s consistent with familial hypercholesterolemia, a medical condition that would indicate immediate medical therapy (e.g. a statin/PCSK9i) but you’re recruiting people who’ve joined an online community that has concluded this isn’t risky so they’re going to opt not to take medication - we live in a rather litigious society and enrolling a cohort of folks considered at high risk denying medical advice is going to be a big liability for any institution to take on.
I chatted about all of this Spencer at the time and watched as it evolved into what is now the “Keto-CTA” cohort (NCT: 05733325). Spencer coordinated with Dave Feldman to work with cardiovascular disease phsyician & researcher, Matt Budoff, at Lundquist, the sponsoring insitution for the cohort. The study ultimately chose to enroll ‘healthy’ particiaptns with an elevated LDL-C (>190mg/dL) considered to be induced by diet and not a genetic familial hypercholesterolemia (note: it’s hard to rule a diet-x-genetic interaction here). Participants in the trial had to have voluntarily consumed a low carbohydrate KD for 24 months and had to have a historical LDL-C lab value showing that levels on the diet increased by at least 50% prior to starting the diet and then elevated while consuming the KD. In order to enroll ‘healthy’ participants, a range of criteria were established: a BMI <30mg/k2 and if the BMI is 25-30mg/kg2, the waist circumference had to fall below 102/88cm for men/women; a HDL-C >60mg/dL and triglycerides <80mg/dL; systolic blood pressures <130mmHg or diastolic <80mmHg; fasting blood glucose <110mg/dL and HbA1c <6.0%; hsCRP <2mg/L; and no prior diagnosis of type 2 diabetes or taking anti-diabetic or lipid lowering medications/supplements. After being recruited online, study participants were enrolled, came in for a baseline visit and then a 12 month follow up visit to allow for blood draws and undergoing cardiac computed tomography angiography (CCTA) . An ECG, vital signs, medical history, physical exam, diet assessments and genetic tests were also done. The study was registered as needing 100 individuals based on a primary endpoint of measuring the % change in total non-calcified coronary plaque volume. To assess adherence to the diet, blood glucose and ketone levels were measured daily using KetoMojo. All in the all, it’s a reasonable study design ( some issues detailed further below) and the cohort is pre-registered to tell us about plaque advancement, a research but non-clinical marker. There are some notable design elements to consider up front.
this is not a randomized controlled trial. it will be challenging to causally say anything.
we don’t have randomization but we also don’t have a ‘healthy’ non-keto comparison cohort, or a group of keto dieters who don’t see an increase in LDL-C (we’d ideally like multiple comparators if we’re trying to say anything specific to keto and specific to keto with high LDL-C). This precludes us from saying too much about what the 1 year change means relative to other groups without having to rely on comparisons with very different cohorts (it is always superior to recruit your own control/comparator cohort that undergoes measures in the same facility with the same machinery and that you can match for but alas, that basically doubles the cost and didn’t happen here).
This is a simple pre-post analysis - changes over time may be an effect of diet, other variables (both measured and unmeasured), and biases like regression to the mean (extreme values at one time point tend to become less extreme over time). Changes over time might also just be technical variability in the CCTA methods.
As a prospective cohort, we will functionally get 1 measure of LDL-C/ApoB at baseline and be able to assess its relationship to the change in measures of plaque over a single year. Compare this with the concern - that years of exposure to high LDL-C/ApoB while consuming a ketogenic diet will exacerbate atherosclerosis and actual cardiovascular events. The design of this cohort is a start but is lacking multiple meaures of blood lipids and long-term followup to the point of assessing cardiovascular disease risk. Plaque measures at baseline or changes over the year are not really clinically validated metrics of cardiovascular disease risk on their own, so we won’t really be alleviating the a priori concern about keto, regardless of the results. What we want to truly know is a tough question to answer but we should be sober about what this study design can even tell us.
This is going to be a highly self-selected population of relatively healthy ketogenic dieters. Elevated LDL-C is accepted as causal in ASCVD but it is far from the only factor that contributes to overall risk, and this cohort has selected for individuals who are healthy on virtually every other metric of risk that’s measured - they dont have diabetes, they’re not smokers, no elevated blood pressure, good metrics on other lipids.
With all that said, let’s get into the main manuscript titled “Plaque Begets Plaque, ApoB Does Not: Longitudinal Data From the KETO-CTA Trial” (I am not sure why we are calling a cohort a trial, it’s a misleading title because it’s a cohort, not a trial).
This paper contains the description of the cohort and the major CCTA outcomes assessing plaque volume data at baseline and 1 year. The cohort was predominantly male (59%) and white (83%) and at baseline, averaged 55yo with ‘normal’ BMIs (avg: 22.5), and LDL-C of 253.7 (the 25th to 75th percentile here is 202-308). The participants had low scores based on the MESA 10yr CVD risk calculator (a composite of age, blood lipids, ethnicity, family history, & coronary artery calcium, diabetes, smoking status and lipid/blood pressure medication usage), consistent with the selection of a pretty healthy cohort apart from their elevated LDL-C. Unfortunately, we don’t get a recruitment flow chart for how many people were screened and didn’t mean the inclusion criteria to get a feel for how many people who self-identify as LMHR actually meet these strict ‘healthy’ criteria. For a diet that’s often presented as a cure-all, it’d be nice to know how many people on it still show measures of less than ideal health.

The cohort followed the diet for an average of 4.5yrs (oddly reported in days down to the 0.1 decimal, which is unlikely to be the unit or level of precision they had for this). paper assessed diet, although they confusingly saying that they collected 3 dietary recalls but also refer to them as dietary records (this is a different thing than a recall and the ASA24 tool does both, i’m not sure what they actually collected). We strangely only get a small subset of the diet data (no calories/protein) but they indicate high fat intakes and low carbohydrate intakes. Only about 1/3 of the fat is reported as saturated fat, which is common in the general population but less typical for many on ketogenic diets. These folks are pretty lean and likely don’t require too many calories but the fat and carbohydrate intakes only add up to 1120 calories. For an average height man (5’7) with a BMI of 22.5, they’d weight about 144lbs and would need somewhere between 1700-2200 kcals to maintain their weight, depending on their activity levels. That’s a big kcal gap to clear with protein alone at 4kcal/g protein - probably underreporting going on but somehow JACC let this diet cohort through without the full results of their dietary analysis.

What about the meat of the paper, it’s primary measures of arterial plaque volume by CCTA? The major outcome we get reported in figure one is non-calcified plaque volume (NCPV) - plaque volume appears to have increased on average, with quite a bit of variability in the cohort with respect to baseline plaque and how much the plaque increased over time. Oddly, there’s no real statistical analysis of the change from baseline to follow-up and we aren’t shown descriptive statistics on the data at baseline, follow-up and the change to characterize the cohort and all of its heterogeniety - it is a big failure on JACC’s editorial board to not have asked for these pretty basic things. You can look at this data and paint a number of stories about it - plaque, on average, seems to worsen! But also, if you squint hard or look to Figure 3, its apparent a number of participants see no change/reductions in NCPV and in the total plaque score - atherosclerosis improves in some people on keto! Before you get too excited about either, remember the design of this study doesn’t let us rule out biology from statistical bias or technical noise and we have no comparator group to say anything about the relative change. I’m not an expert in all of the technical aspects of CCTA but the several studies I can find - even in the control arm of lifestyle interventions for very high risk individual & in both SGLT2i and non-SGLT2i users- demonstrate a substantial distribution of gain and ‘regression’ of plaque from baseline to follow up - any individual arm of these would be uninterpretable without the comparator group that starts to show whether there looks to be a relative protective effect of a therapy. Ive seen folks online looking at the cohort and saying, omg keto causes some people’s plaque to regress! - to be intellectually honest (but still wrong), you’d have to look at the whole body of literature and conclude the standard american diet in everyday high risk patients causes plaque to regress in some consumers too.
{kind=link}
One key thing about this Figure 1 data that is the subject of much of the online commentary is that the units here are absolute measures of NCPV (mm3) but the preregistered outcome on clinicaltrials.gov (ostensibly the measure the cohort was powered around) was the percent change in the NCPV. This is major JACC Editorial Board failing #2 (at least). The percent change in NCPV was also stated in their published protocol paper.

The lead author confirmed the average NCPV change in terms of an absolute value as 18.8mm3. Interpreting this absolute number is challenging given the substantial spread but as Nicola Guess points out in her review of the study, this is on the order of what is observed in studies following individuals over ~3yrs who have substantial artery disease and metabolic syndrome. That’s still not the % change though. We can guestimate that it would be pretty substantial given that the mediam NCPV was 44. That’s a pretty high percentage change on average (again there’s variability and this could be driven by outliers). For a comparison, the control arm of the EPA/Vascepa intervention study saw a 9% increase in NCPV in much higher risk patients over 18months.

The variability seen in the cohort, the lack of real stats done on the baseline vs follow up, and the switching of primary outcome units made me find this whole paper a bit sketchy and want to stop reading entirely. I’m not surprised on Twitter that the authors are highlighting the variability angle, seemingly trying to distract from doing what they a priori said they planned to do. It’s fine to have a cohort that ends up being way more heterogeneous than you thought but you should report your analyses nonetheless. This variability got me interested in the power calculations in the protocol paper that ultimately led to deciding they’d only need 100 particiapnts. Confusingly, the power calculation is stated as being based on an absolute change metric of 7mm3 with a SD of 64.5mm3. So the authors pre-registered a primary outcome that states they’ll look at percent change but then powered based on an absolute change (?).

This all didn’t sound right to me - a 7mm3 change with a SD of 64.5 sounded to me like you’d need a lot higher than 100 people, even for 80% power. I decided to model the power calculations in Gpower. The authors don’t say what stats test they’re going to use to detect a change from baseline to follow-up but i assumed a Wilcoxon signed-rank test for matched pairs, given that we don’t assume normal distributions here with all the variability and this is a pre-post test in the same participants. Putting in the mean difference of 7 and SD of 64.5 with 80% power and an alpha of 5%, it is estimated that one would need a sample size of 700, not 90 (they assumed 10% dropout), to see a difference between the baseline and follow up. As the authors say in their power analysis section of the protocol paper, most of this is guess work but it’s not clear to me how this was powered and the protocol paper does not lend confidence that this was a coherent effort. (Note: I played around with power calculation metrics beyond just the screenshot below & its hard to come up with a N below 500, so the high number here isn’t an artefact of choosing the Wilcoxon test).

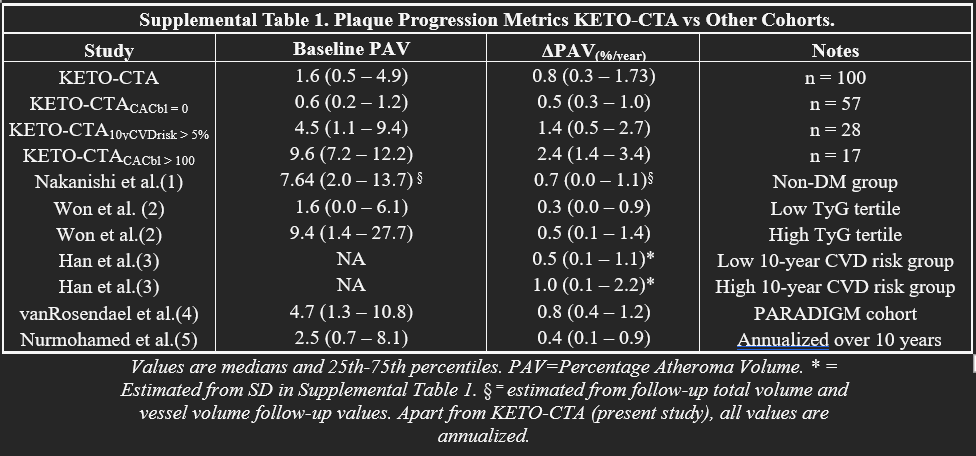
Moving on to other outcomes, in the supplemental material, we get additional data comparing the percent atheroma volume, a different CCTA derived metric that looks at total plaque volume divided by the vessel volume x 100. We can see this cohort alongside several others for comparisons with their baseline and absolute changes in volume. If you look at the percent change, it looks to be pretty high (this isn’t a perfect mathematical way to do it given the heterogeneity in the data but a delta PAV of 0.8 is 50% of the baseline 1.6 PAV). For the other cohorts, that relative change is much lower. Although the absolute PAV puts these folks in a low risk category (expected), this rate of change is concerning and would be even moreso if we saw this rate per year for several years out (alas we left with only 1 year data). For additional context, in his risk patients in both a DASH lifestyle arm and control intervention, participants saw about a 1% increase in PAV over a year.
The rest of the paper goes on to look at predictors of plaque volume metrics. The authors are making more of this than I care to on social media, particularly around the issue of ApoB not being a major predictor of plaque change. There’s a bunch of reasons this isn’t too surprising:
This is a cohort already selected for folks with hypercholesterolemia. In the selection criteria, you’ve already reduced your ability to compare low vs moderate vs high and very high lipoprotein particle metrics like ApoB by selecting only those with already elevated levels. In a population with such heterogeneous baseline plaque levels, it is wholly unsurprising to me that the risk factor you narrowed the range of to the extreme end of a distribution via your inclusion criteria isn’t going to have substantial explanatory value. This selecting for high ApoB/LDL-C at baseline also increases the risk of collider bias, explained well here by Dr Georgakis.
This cohort was never designed or powered to predict factors associated with change in plaque volume over time. We are relying on an underpowered cohort with only 2 time points where ApoB is measured to try to predict plaque change. Most individuals had an increase or minimal change in NCPV, whereas the ApoB levels drop from the baseline to the 1 yr follow up visit - of course it won’t have much explanatory value. None of this tells us much about whether sustained elevations in ApoB will increase plaque progression because we don’t have the sample size, variation in either variable, and/or long-term follow up with repeated measures of ApoB or appropriate comparator cohorts. I’d actually love a full 2-x-2 cohort of Keto vs Not Keto and Elevated LDL-C vs not elevated LDL-C, to start to tease out relative and interactive effects of the keto diet and lipoproteins on plaque progression - alas, we don’t have that here and probably won’t have it any time soon.
I have concerns about doing a bunch of linear modeling with data with this kind of spread that’s probably violating a bunch of normality assumptions - I would have bet at baseline the plaque and ApoB data probably aren’t normally distributed and would need to be log transformed (the NCPV data in the EVAPORATE trial was log transformed, for example)..
I’m not a CCTA expert but I increasingly have concerns about the methods to measure plaque in this low risk cohort, particularly using AI-assisted quantification that looks to be predominantly validated in high risk cohorts where the signal to noise ratio is much higher. The potential for technical variation/error in plaque assessment here looks to be high - I’d be curious to see more cardiology/CCTA comment on this issue.
The only thing that really emerges is that folks with baseline plaque have more plaque progression that isn’t predicted by most existing risk factors. The authors use measures of baseline plaque to predict the change in plaque, which itself is calculated from a metric involving baseline plaque level - it’s circuitous and lends itself to spurious findings, so this predictive’ capacity isn’t surprising (yet makes it into the title..).
The notion that single ApoB measures aren’t massively predict of plaque isn’t super novel. For comparison, we know there’s considerable variability in the degree of plaque even in folks with FH, and a weak relationship between a single measure of LDL-C and plaque, while many other factors show quite stronger associations. Atherosclerosis is a multifactorial disease and 2 things can be true: modifying cumulative LDL-C can modify disease risk but other factors can matter as well.
We should seriously question why the author group, inclusive of large advocates for the keto diet and with several intellectual/financial ties to the topic, are so keen to ditch the pre-specified analytical plan for their cohort in favor of exploratory analyses they were never powered to explore, and then highlight this as the major finding of their paper in its title and all over social media. How the JACC editors alowed this is beyond me and its a total editorial failure.

I’m curious what’s next for this cohort and if it will continue - truthfully, I’m not sure that it should. I had doubts about the ethics of this cohort in the beginning, largely centered around Feldman, the originator of the lipid triad hypothesis. Feldman started the Citizen Science Foundation that is listed as a major collaborator of the study and had a role in the funding, design, recruitment and analysis of the study and is on the major papers from it. Think about this - the guy who posited a theory and created a massive online community around the idea that high LDL-C isn’t harmful if you have other wise low TG and high HDL-C on low carb is involved in every aspect of a study designed to test his hypothesis and fundraising/recruiting from his own online community. This potential for bias decreases trust in every aspect of the study results and this bias should’ve been sequested by Lundquist’s IRB. It is generally shocking to me that Lundquist served as the hosting institution for a cohort of folks where if one person has a heart attack, the families could sue like crazy because the informed consent didn’t convey potential harms of enrolling in the study and foregoing lipid lowering therapy (an exclusion criteria of the study). This ethical wonkyness of the study now feels even worse as we’re seeing its results which is a mess of heterogeneity - participants in the study and the broader keto community are seeing both researchers/doctors interpreting that the study shows rapid progression in at least a subset of participants on the ketogenic diet as well as the study’s own authors out here saying the lipid hypothesis has crumbed

I have tons of questions - are these results getting communicated back to patients? is Lundquist providing care or ensuring that they are getting offered care? The potential for medical negligence here is high, for research that doesn’t clearly benefit its participants and is now being spun in a way that could actually harm them. I’ll be curious to see if Lundquist allows this to continue.